Introduction
Site Occupation Method
In molecular dynamics (MD) simulations, vacancy positions are not tracked explicitly. Instead, they must be identified implicitly by assigning each atom to its corresponding lattice site and then locating the sites that remain unoccupied. Consequently, studying vacancy-mediated diffusion requires a highly accurate method for this site assignment.
This assignment cannot be based on simple proximity alone. A more robust criterion is needed that considers the atom’s position relative to the transition state (TS), which is revealed by the force acting on the atom.
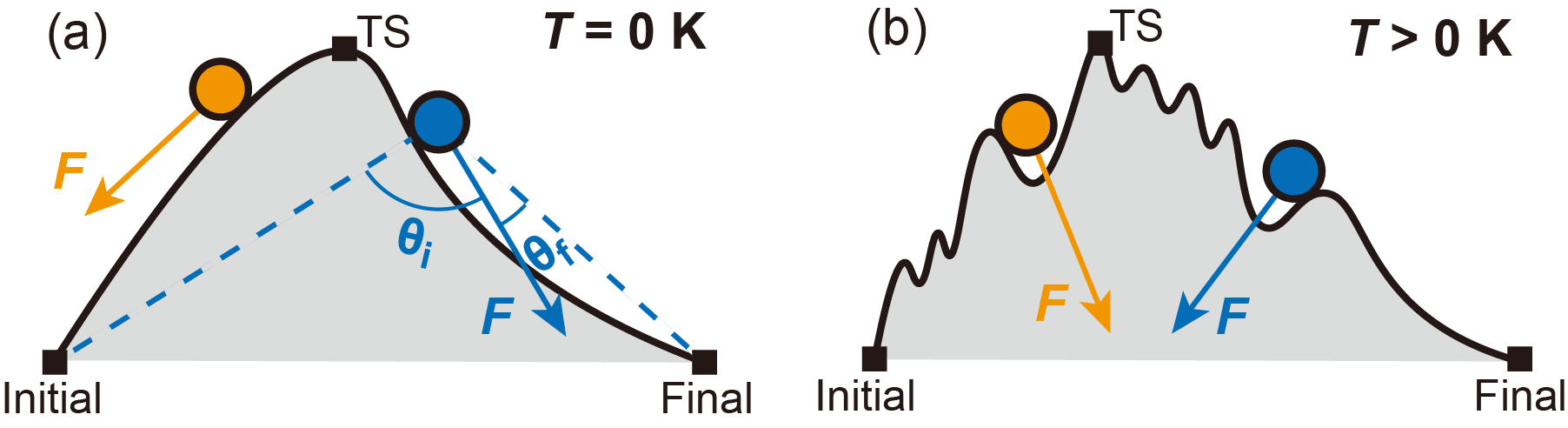
Figure 1. Force on an atom (a) in an ideal 0 K system and (b) in a finite-temperature MD simulation.
As shown in Figure 1(a), in an ideal 0 K system, the force vector on an atom points directly toward the center of its occupied lattice site. In a finite-temperature MD simulation, however, this force vector is heavily perturbed by random thermal fluctuations (Figure 1(b)), making it difficult to use the instantaneous force for reliable site assignment.
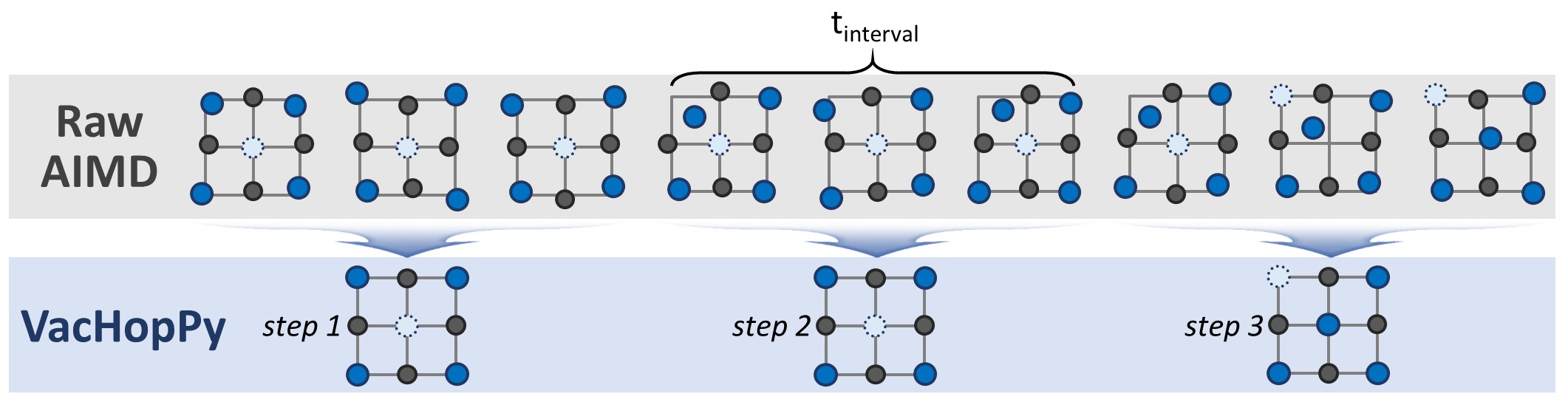
Figure 2. The coarse-graining approach for noise cancellation.
To overcome this challenge, VacHopPy employs a coarse-graining approach. Instead of analyzing the raw trajectory, it averages atomic positions and forces over a defined time interval (t_interval), as illustrated in Figure 2. Because thermal fluctuations are random, this time-averaging process effectively cancels out the noise, revealing the underlying, physically meaningful force.
Theoretically, the ideal t_interval corresponds to the inverse of the characteristic atomic vibration frequency (\(\nu^*\)). For typical solids, this frequency is on the order of 10 THz, which suggests an optimal t_interval of approximately 0.1 ps.
This site allocation method is particularly crucial for materials with complex crystal structures, such as those containing inequivalent lattice sites (e.g., monoclinic HfO₂). In these systems, the transition state is often not located at the geometric midpoint between two sites. A simple proximity-based assignment would therefore be unreliable, making VacHopPy’s force-based criterion essential for an accurate analysis.
Effective Hopping Parameters
In ab initio calculations, such as the nudged elastic band (NEB) method, hopping parameters are typically determined for a specific, predefined migration path. Consequently, in a system with multiple distinct migration pathways, one obtains a collection of different hopping parameter sets, each corresponding to a unique path.
However, to compare these computational results with experimental data or to use them in continuum-scale models, a single, representative set of parameters that encapsulates the overall diffusion behavior is required. We term this representative set the effective hopping parameters, which serve as ready-to-use inputs for continuum-level analysis.
The effective hopping parameters self-consistently satisfy the following fundamental diffusion equations:
For a more detailed theoretical background, please refer to this paper.
The key effective hoppping parameters extractable with VacHopPy are summarized below.
Symbol |
Parameter |
Unit |
|---|---|---|
\(D\) |
Vacancy diffusivity |
m²/s |
\(\tau\) |
Vacancy residence time |
ps |
\(f\) |
Correlation factor |
- |
\(E_a\) |
Hopping barrier |
eV |
\(a\) |
Hopping distance |
Å |
\(z\) |
Coordination number |
- |
\(\nu\) |
Attempt frequency |
THz |
\(\nu^*\) |
Atomic vibration frequency |
THz |
All hopping parameters extracted by VacHopPy, such as those summarized above, are effective values. This means they represent the overall, averaged behavior of vacancy diffusion across all possible migration paths within the system. For instance, the calculated effective hopping barrier (\(E_a\)) is not tied to a single NEB path but reflects the system-wide average barrier influencing diffusion.
For other extractable parameters, please refer to this section.
Note
A Note on Key Terminology
It is important to distinguish between two pairs of commonly confused terms:
Attempt Frequency (\(\nu\)) vs. Atomic Vibration Frequency (\(\nu\)*)
While often used interchangeably, these are distinct quantities. The atomic vibration frequency (\(\nu^*\)) is a result of all phonon modes in the system, whereas the attempt frequency (\(\nu\)) is typically associated with a single phonon branch relevant to a specific hopping path.
Hopping Barrier vs. Diffusion Barrier
The hopping barrier is the direct energy cost for a species to make a single hop. The diffusion barrier, in contrast, is the macroscopic activation energy for the overall diffusion process, which includes both the hopping barrier and contributions from jump correlation effects.
For example, the activation energy of the tracer diffusivity (\(D\)) corresponds to the diffusion barrier. The activation energy for quantities related to individual jumps—such as random-walk diffusivity (\(D_{rand}\)), residence time (\(\tau\)), or the hopping rate (\(\Gamma\))—corresponds to the hopping barrier.
VacHopPy explicitly differentiates between these concepts to provide physically accurate parameters.
Additional Tools
In addition to the primary parameter extraction, VacHopPy also provides several secondary tools for more in–depth analysis:
Vacancy Trajectory Visualization
Visualize the movement and pathways of vacancies over time.
MSD-based Diffusivity Calculation
Calculate atomic diffusivity using mean squared displacement (MSD) analysis.
Structural Evolution Tracking
Identify and track the evolution of the material’s structure throughout the simulation.